What is aggregate-data modelling?
admixr2 fits pharmacometric PK/PD models to
summary-level data. For each clinical study you
supply:
- E — observed mean vector (one entry per observation time)
- V — observed covariance matrix (or variance vector)
- n — sample size
- times — observation time points
- ev — dosing event table
The estimators match E and V against their model-predicted counterparts and return a standard nlmixr2 fit object. This lets you apply established nlmixr2 models to aggregate statistics from publications or internal data summaries where individual records are unavailable.
Four estimators are available:
| Estimator | est = |
Control function | Approach |
|---|---|---|---|
| First-Order | "adfo" |
adfoControl() |
First-order Taylor expansion at η = 0; one rxSolve per NLL eval; fastest |
| Monte Carlo | "admc" |
admControl() |
Sample average over η; asymptotically exact |
| Gauss-Hermite | "adgh" |
adghControl() |
Deterministic quadrature over η; noise-free, unbiased at any IIV |
| Iterative Reweighting MC | "adirmc" |
adirmcControl() |
Proposals fixed per phase; inner loop needs no new rxSolve calls |
adfo is the natural starting point for model screening
and initial estimates. admc is the workhorse for standard
PK models. adgh is a noise-free alternative to
admc for models with up to ~4 etas. adirmc is
preferred for complex ODE systems with expensive solves,
high-dimensional IIV, or poor starting values. See
vignette("estimator-comparison", package = "admixr2") for a
detailed comparison.
The examplomycin dataset
examplomycin ships with admixr2: 500 simulated subjects
from a two-compartment PK model with first-order absorption (100 mg oral
dose, sampled at 0.1, 0.25, 0.5, 1, 2, 3, 5, 8, and 12 h). True
parameters: CL = 5 L/h, V1 = 10 L, V2 = 30 L, Q = 10 L/h, ka = 1 h⁻¹;
IIV = 0.3 (SD on log scale) for all parameters; proportional error SD =
0.2.
library(admixr2)
library(rxode2)
library(nlmixr2)
data("examplomycin")
head(examplomycin[examplomycin$EVID == 0, c("ID", "TIME", "DV")], 9)
#> ID TIME DV
#> 2 460 0.10 0.752
#> 3 460 0.25 1.932
#> 4 460 0.50 3.694
#> 5 460 1.00 3.479
#> 6 460 2.00 4.003
#> 7 460 3.00 3.825
#> 8 460 5.00 1.756
#> 9 460 8.00 1.155
#> 10 460 12.00 0.742Computing aggregate statistics
Reshape individual records into a subjects × times matrix, then compute E and V:
obs <- examplomycin[examplomycin$EVID == 0, ]
obs <- obs[order(obs$ID, obs$TIME), ]
times <- sort(unique(obs$TIME))
ids <- unique(obs$ID)
n <- length(ids) # 500
dv_mat <- matrix(NA_real_, nrow = n, ncol = length(times))
for (i in seq_along(ids)) {
sub <- obs[obs$ID == ids[i], ]
dv_mat[i, ] <- sub$DV[order(sub$TIME)]
}
E <- colMeans(dv_mat)
V <- cov.wt(dv_mat, method = "ML")$cov
round(E, 2)
#> [1] 0.97 1.94 2.79 3.02 2.26 1.65 1.06 0.75 0.51V is the 9×9 sample covariance matrix. Its off-diagonal
entries capture within-subject correlation across time; using the full
matrix (method = "cov") typically tightens parameter
estimates compared to the diagonal-only approximation
(method = "var"). admixr2 auto-detects the
method from the structure of V.
Model definition
Models use standard nlmixr2 syntax with mu-referenced log-scale parameters:
pk_model <- function() {
ini({
tcl <- log(5) ; label("Log clearance (L/hr)")
tv1 <- log(10) ; label("Log central volume (L)")
tv2 <- log(30) ; label("Log peripheral volume (L)")
tq <- log(10) ; label("Log inter-compartmental CL (L/hr)")
tka <- log(1) ; label("Log absorption rate constant (1/hr)")
prop.sd <- c(0, 0.2); label("Proportional residual error SD")
eta.cl ~ 0.09
eta.v1 ~ 0.09
eta.v2 ~ 0.09
eta.q ~ 0.09
eta.ka ~ 0.09
})
model({
cl <- exp(tcl + eta.cl)
v1 <- exp(tv1 + eta.v1)
v2 <- exp(tv2 + eta.v2)
q <- exp(tq + eta.q)
ka <- exp(tka + eta.ka)
d/dt(depot) <- -ka * depot
d/dt(central) <- ka * depot - (cl/v1 + q/v1) * central + (q/v2) * peripheral
d/dt(peripheral) <- (q/v1) * central - (q/v2) * peripheral
cp <- central / v1
cp ~ prop(prop.sd)
})
}Writing each parameter as exp(tcl + eta.cl) is called
mu-referencing: the structural fixed effect and its
random effect enter additively on the log scale. admixr2
exploits this pairing to compute analytical gradients via sensitivity
equations. See the Advanced
usage vignette for details, including how parameters without a
random effect are handled.
Fitting
Pass one or more named studies to admControl():
fit <- nlmixr2(
pk_model, admData(), est = "admc",
control = admControl(
studies = list(examplomycin = study),
n_sim = 5000L,
cov_n_sim = 10000L,
maxeval = 300L,
seed = 1L
)
)
#> [====|====|====|====|====|====|====|====|====|====] 0:00:07Inspecting the fit
print(fit)
#> ── nlmixr² admc ──
#>
#> OBJF AIC BIC Log-likelihood
#> admc -3690.835 -3668.835 -3598.305 1845.418
#>
#> ── Time (sec fit$time): ──
#>
#> optimize covariance elapsed
#> 1 38.767 9.275 48.042
#>
#> ── Population Parameters (fit$parFixed or fit$parFixedDf): ──
#>
#> Parameter Est. SE %RSE
#> tcl Log clearance (L/hr) 1.601 0.01635 1.021
#> tv1 Log central volume (L) 2.314 0.08719 3.768
#> tv2 Log peripheral volume (L) 3.402 0.04007 1.178
#> tq Log inter-compartmental CL (L/hr) 2.285 0.02132 0.9332
#> tka Log absorption rate constant (1/hr) 0.02423 0.08198 338.4
#> prop.sd Proportional residual error SD 0.1984
#> Back-transformed(95%CI) BSV(CV%) Shrink(SD)%
#> tcl 4.958 (4.802, 5.12) 32.8
#> tv1 10.12 (8.528, 12) 33.8
#> tv2 30.03 (27.76, 32.48) 32.0
#> tq 9.822 (9.42, 10.24) 33.4
#> tka 1.025 (0.8725, 1.203) 31.2
#> prop.sd 0.1984
#>
#> Covariance Type (fit$covMethod): r
#> No correlations in between subject variability (BSV) matrix
#> Full BSV covariance (fit$omega) or correlation (fit$omegaR; diagonals=SDs)
#> Distribution stats (mean/skewness/kurtosis/p-value) available in fit$shrink
#> Censoring (fit$censInformation): No censoring
#> Minimization message (fit$message):
#> NLOPT_XTOL_REACHED: Optimization stopped because xtol_rel or xtol_abs (above) was reached.Key entries in fit$env$admExtra:
fit$objective # -2 log-likelihood
#> [1] -3690.835
fit$env$admExtra$struct # structural parameters (log scale)
#> tcl tv1 tv2 tq tka
#> 1.60106954 2.31424059 3.40223700 2.28459888 0.02422601
fit$env$admExtra$omega # estimated Omega matrix
#> [,1] [,2] [,3] [,4] [,5]
#> [1,] 0.102159 0.0000000 0.00000000 0.000000 0.00000000
#> [2,] 0.000000 0.1080019 0.00000000 0.000000 0.00000000
#> [3,] 0.000000 0.0000000 0.09747445 0.000000 0.00000000
#> [4,] 0.000000 0.0000000 0.00000000 0.105588 0.00000000
#> [5,] 0.000000 0.0000000 0.00000000 0.000000 0.09281045
fit$env$admExtra$sigma_var # residual variance(s)
#> prop.sd
#> 0.03937635
logLik(fit)
#> 'log Lik.' 1845.418 (df=11)
AIC(fit)
#> [1] -3668.835Diagnostic plots
plot() produces up to four panel types and returns them
as a named list of ggplot2 objects:
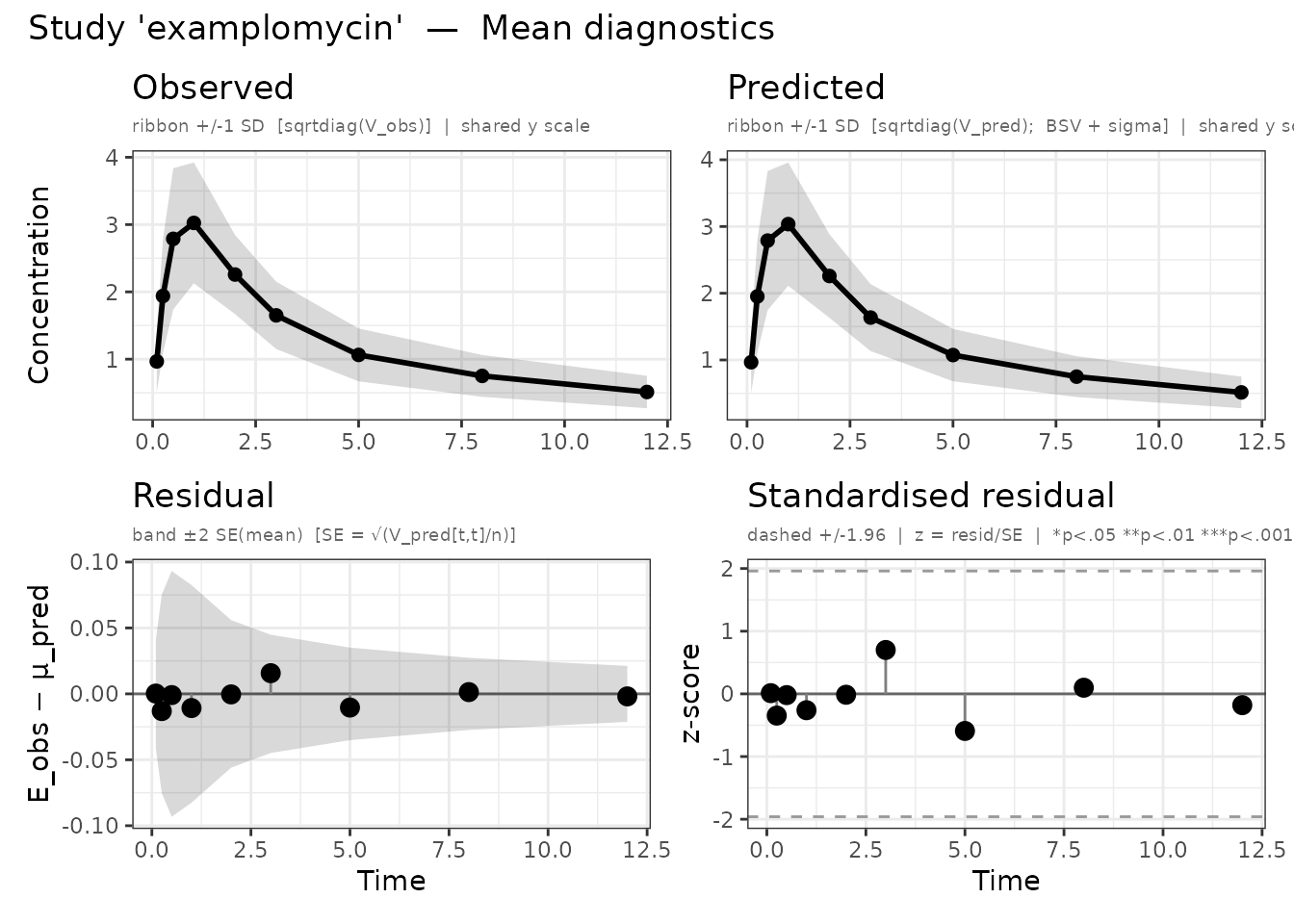
Left: observed vs predicted mean with residuals. Right: NLL convergence trace.
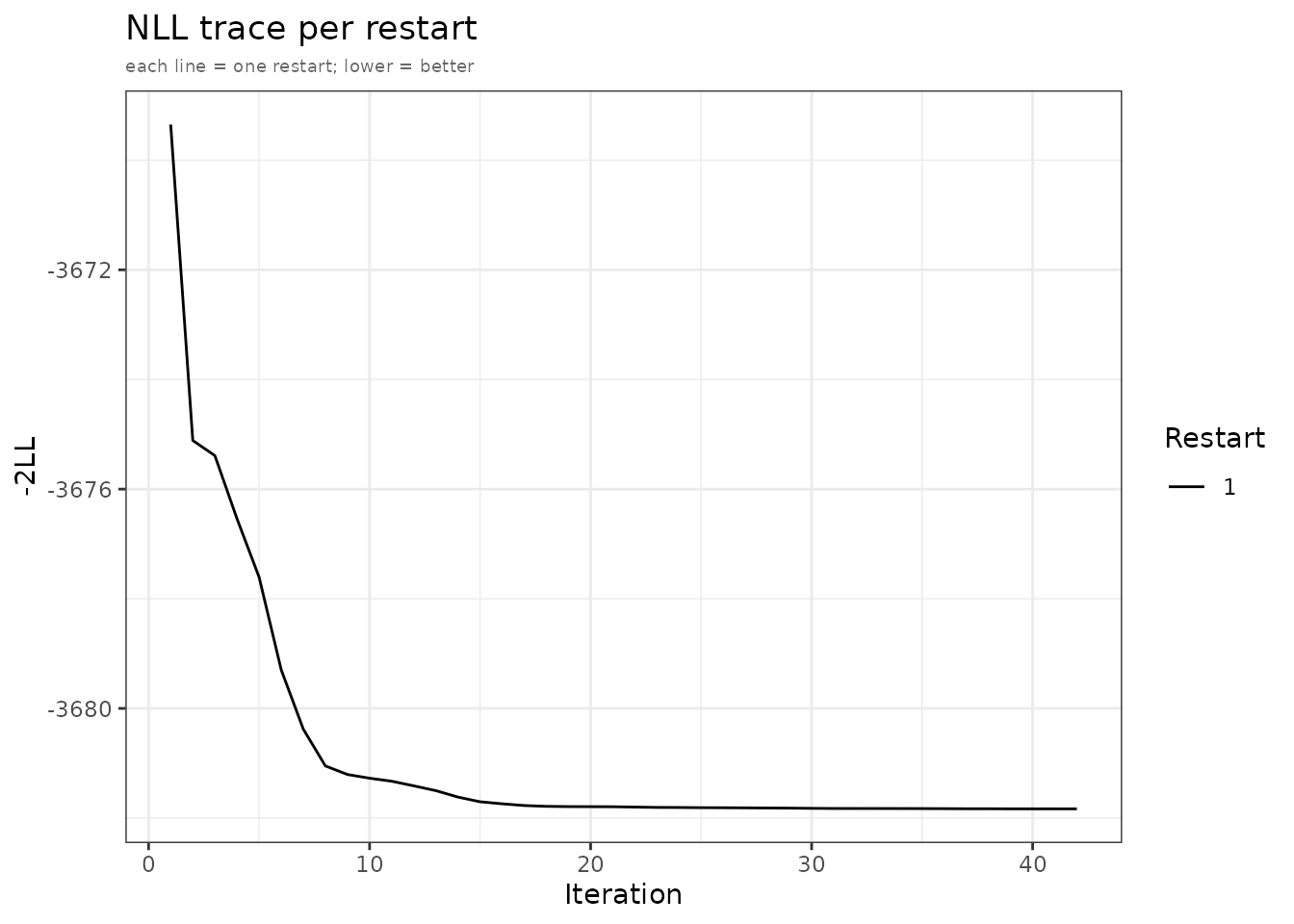
Left: observed vs predicted mean with residuals. Right: NLL convergence trace.
For a detailed walkthrough of all four panel types and customisation
options, see
vignette("diagnostic-plots", package = "admixr2").